서로 다른 사람이 실험해서 얻은 pdb 파일은 좌표가 제각각이라 구조를 비교를 하기 전에 먼저 정렬을 먼저 해야 합니다.
pymol은 RMSD가 최소가 되도록 구조 정렬을 해줍니다. RMSD보다는 TMscore가 더 장점이 있긴 하지만, TMscore와 TMalign으로 정렬을 하는 건 조금 불편합니다. 그래도 TMscore나 TMalign도 최근엔 구조 정렬 결과를 결과 파일로 출력해서 pymol에서 읽어올 수 있도록 하는 기능을 지원합니다.
먼저 pymol *.pdb 로 EGFR에 대한 구조들을 읽어오겠습니다. 제일 마지막에 읽어 들인 구조에 맞춰서 화면이 출력되었습니다.

pymol에서 구조 정렬하는 옵션으로 align과 alignto 가 있습니다. align은 특정 구조를 reference구조에 대해서 정렬하라는 명령어입니다. alignto는 읽어 들인 모든 구조에 대해서 특정 reference에 대해서 정렬하라는 명령어입니다.
각각
align A,B_ref
align A_ref
같은 방식으로 실행합니다.
정확한 차이는 모르겠지만, align과 alignto를 사용한 결과는 다릅니다.
다중구조일 때라도 귀찮아도 align을 사용하는 것을 추천합니다.
(좌) alignto 실행 결과, (우) align 실행결과
서로 RMSD 값이 다르게 나옵니다.
align
align 1M14, 5FED
align 2GS2, 5FED
align 2ITY, 5FED
align 2RGP, 5FED
align 3VJO, 5FED
align 4TKS, 5FED를 순서대로 실행한 결과
PyMOL>align 1M14, 5FED
Match: read scoring matrix.
Match: assigning 324 x 316 pairwise scores.
MatchAlign: aligning residues (324 vs 316)...
MatchAlign: score 1466.500
ExecutiveAlign: 2309 atoms aligned.
ExecutiveRMS: 88 atoms rejected during cycle 1 (RMSD=9.98).
ExecutiveRMS: 60 atoms rejected during cycle 2 (RMSD=1.40).
ExecutiveRMS: 173 atoms rejected during cycle 3 (RMSD=0.69).
ExecutiveRMS: 143 atoms rejected during cycle 4 (RMSD=0.39).
ExecutiveRMS: 110 atoms rejected during cycle 5 (RMSD=0.28).
Executive: RMSD = 0.235 (1735 to 1735 atoms)
PyMOL>align 2GS2, 5FED
Match: read scoring matrix.
Match: assigning 329 x 316 pairwise scores.
MatchAlign: aligning residues (329 vs 316)...
MatchAlign: score 1469.500
ExecutiveAlign: 2308 atoms aligned.
ExecutiveRMS: 150 atoms rejected during cycle 1 (RMSD=0.92).
ExecutiveRMS: 145 atoms rejected during cycle 2 (RMSD=0.51).
ExecutiveRMS: 122 atoms rejected during cycle 3 (RMSD=0.38).
ExecutiveRMS: 75 atoms rejected during cycle 4 (RMSD=0.32).
ExecutiveRMS: 40 atoms rejected during cycle 5 (RMSD=0.29).
Executive: RMSD = 0.280 (1776 to 1776 atoms)
PyMOL>align 2ITY, 5FED
Match: read scoring matrix.
Match: assigning 320 x 316 pairwise scores.
MatchAlign: aligning residues (320 vs 316)...
MatchAlign: score 1422.000
ExecutiveAlign: 2252 atoms aligned.
ExecutiveRMS: 53 atoms rejected during cycle 1 (RMSD=1.58).
ExecutiveRMS: 141 atoms rejected during cycle 2 (RMSD=0.69).
ExecutiveRMS: 117 atoms rejected during cycle 3 (RMSD=0.44).
ExecutiveRMS: 81 atoms rejected during cycle 4 (RMSD=0.36).
ExecutiveRMS: 46 atoms rejected during cycle 5 (RMSD=0.33).
Executive: RMSD = 0.316 (1814 to 1814 atoms)
PyMOL>align 2RGP, 5FED
Match: read scoring matrix.
Match: assigning 372 x 316 pairwise scores.
MatchAlign: aligning residues (372 vs 316)...
MatchAlign: score 1338.500
ExecutiveAlign: 2186 atoms aligned.
ExecutiveRMS: 120 atoms rejected during cycle 1 (RMSD=3.43).
ExecutiveRMS: 113 atoms rejected during cycle 2 (RMSD=2.00).
ExecutiveRMS: 92 atoms rejected during cycle 3 (RMSD=1.63).
ExecutiveRMS: 95 atoms rejected during cycle 4 (RMSD=1.46).
ExecutiveRMS: 98 atoms rejected during cycle 5 (RMSD=1.29).
Executive: RMSD = 1.118 (1668 to 1668 atoms)
PyMOL>align 3VJO, 5FED
Match: read scoring matrix.
Match: assigning 308 x 316 pairwise scores.
MatchAlign: aligning residues (308 vs 316)...
MatchAlign: score 1473.000
ExecutiveAlign: 2312 atoms aligned.
ExecutiveRMS: 133 atoms rejected during cycle 1 (RMSD=0.93).
ExecutiveRMS: 149 atoms rejected during cycle 2 (RMSD=0.50).
ExecutiveRMS: 112 atoms rejected during cycle 3 (RMSD=0.36).
ExecutiveRMS: 74 atoms rejected during cycle 4 (RMSD=0.31).
ExecutiveRMS: 47 atoms rejected during cycle 5 (RMSD=0.29).
Executive: RMSD = 0.277 (1797 to 1797 atoms)
PyMOL>align 4TKS, 5FED
Match: read scoring matrix.
Match: assigning 307 x 316 pairwise scores.
MatchAlign: aligning residues (307 vs 316)...
MatchAlign: score 1468.500
ExecutiveAlign: 2312 atoms aligned.
ExecutiveRMS: 146 atoms rejected during cycle 1 (RMSD=0.97).
ExecutiveRMS: 138 atoms rejected during cycle 2 (RMSD=0.50).
ExecutiveRMS: 103 atoms rejected during cycle 3 (RMSD=0.36).
ExecutiveRMS: 79 atoms rejected during cycle 4 (RMSD=0.31).
ExecutiveRMS: 60 atoms rejected during cycle 5 (RMSD=0.29).
Executive: RMSD = 0.272 (1786 to 1786 atoms)
이렇게 변환을 한 다음에 변환된 구조를 pdb로 다시 저장해 두고 싶다면,
File -> Export Molecule을 선택합니다. (File을 클릭하면 스크린숏이 안 찍히네요.)
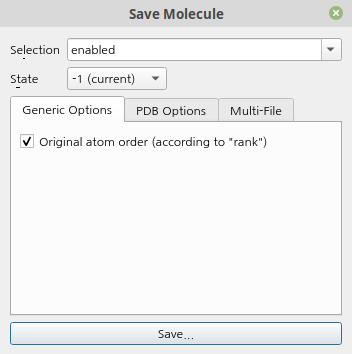
다음과 같은 화면이 나옵니다. 저장할 pdb를 선택할 수 있는데, 여러 pdb 구조들을 한 파일에 저장할 수도 있습니다.
체크 옵션으로 Original atom order가 있는데, 체크를 해주는 편이 좋습니다.
1M 14의 경우 HETATM이 ATOM보다 residue number가 더 앞에 오는데, 이것을 체크 안 하고 저장하면, HETATM이 제일 먼저 저장되어버립니다.
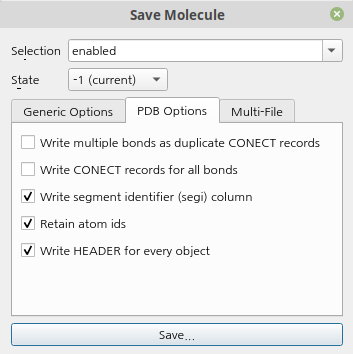
2번째 탭에선 CONECT에 bond 정보를 기록할지 선택할 수 있습니다.
pdb 파일에서 ATOM으로 표기되는 20가지 기본 아미노산은 기본적으로 CONECT를 기록하지 않습니다.
기록하고 싶다면 2번째 항목을 체크합니다.
PDB 사이트에서 다운로드한 pdb 파일은, 기본적으로 bond type (single, double, triple)을 기록하지 않습니다. 그래서 pdb에 붙은 ligand의 aromatic ring이 잘못 파싱 되는 경우가 상당히 많습니다. pymol도 완벽하게 해주지 못합니다. 이걸 제대로 하고 싶으면, pdb 사이트에서 제공하는 cif 파일을 다운로드하여야 합니다.
공식적으로는 bond type을 기록하지 못한다고 해도, 많은 프로그램들이 CONECT에 숫자를 2번 적으면 double, 3번 적으면 triple로 식별하는 기능을 지원하고 있습니다.
다만, aromatic의 경우는 조심해야 합니다. pymol의 경우는 4번 적힌 경우가 aromatic이지만, 다른 프로그램에선 4번 적힌 경우를 4중 결합으로 인식해버립니다. pdb 파일에서 aromatic form을 쓰는 건 좀 어렵고, Kekule form 형태로 사용하여야 합니다. 아무튼 여기선 체크하지 않겠습니다.
아마도 pymol에서 특정 영역을 지우거나 더하지 않았다면 retain atom ids는 영향이 없을 것 같습니다.
특정 영역을 지우고 index를 초기화하고 싶다면 체크를 해제합니다.
HEADER는 별로 쓸 일은 없을 것 같지만, 체크했습니다.
save 파일을 누르면, 파일명과 확장자를 선택하고 저장할 수 있습니다.
원하는 확장자를 선택합니다. 저는 pdb를 선택했습니다. 요즘 cif도 많이 쓰는 것 같네요.
사실 저는 이런 방법을 별로 선호하지 않습니다. 왜냐하면, ATOM, HETATM 이외에 다른 섹션들이 많이 사라지기 때문입니다.
missing residue에 대한 정보가 필요한 경우는 좀 곤란합니다. 물론 복사해서 붙여 넣으면 됩니다만...
'Drug > Computer-Aided Drug Discovery' 카테고리의 다른 글
| fragment 기반 약물 가상탐색 (0) | 2022.02.03 |
|---|---|
| PDB 파싱: CONECT 채워넣기 예제 (0) | 2022.01.29 |
| pymol 사용법: protein-ligand interaction 보기 (0) | 2022.01.24 |
| pymol 사용법: pymol open source 설치 (0) | 2022.01.24 |
| cheminformatics 툴킷: rd_filters (3) | 2022.01.18 |